Reading atoms from configuration files
MDMC enables reading atomic configurations and topologies (if included) from configuration files.
Atomic configurations are read through ASE, so any format supported by ASE is available.
The simplest way to do this is by passing the file to the MDMC.readers.configurations.read function. If the file format is supported, MDMC will automatically use the file extension to read it correctly. In this instance, our example paracetamol.cif file is read as a list of atoms, which we then put into a Molecule object.
[1]:
from MDMC.readers import configurations
atoms = configurations.read('data/Paracetamol.cif')
# The return type of read is a list of atoms
print('The return type is: {}'.format(type(atoms)))
The return type is: <class 'list'>
[2]:
# We can use this to create a Molecule
from MDMC.MD import Molecule
paracetamol = Molecule(atoms=atoms)
The paracetamol Molecule read from the CIF file can be visualized using the view function:
[4]:
from MDMC.gui import view
view(paracetamol, viewer='ASE')
From this it appears that the configuration and topology has been correctly read; however, MDMC cannot automatically determine if certain atoms or bonded interactions are equivalent. For example, all three hydrogens in the methyl group are equivalent, as are the bonds, bond angles and dihedrals these hydrogens are involved in. In the case of paracetamol, there are 20 bonds in total, however only 12 of them are unique.
[ ]:
# To see all of the Bond interactions, we can filter the paracetamol interactions by name
bonds = list(filter(lambda x: x.name == 'Bond', paracetamol.interactions))
# There are 20 bonds in total
print('Number of bonds: {}'.format(len(bonds)))
# We can cast the list of bonds to a set to see the number of unique bonds - in this case it is still 20
unique_bonds = set(bonds)
print('Number of unique bonds: {}'.format(len(unique_bonds)))
Specifying equivalent atoms
There are two methods for specifying which atoms are equivalent; these can be seen by viewing the documentation relevant to the file format of the configuration file, which can be accessed by passing docstring=True when calling read. For example, with a CIF file:
[ ]:
configurations.read('Paracetamol.cif', docstring=True)
From this, it is possible to define equivalent atoms either by specifying the name of each Atom or the atom_type of each Atom.
These names or atom types must be passed in the same order as the atoms are defined within the CIF file (i.e. in the _atom_site_label identifier).
Equivalence specified by atom_types
As with all atom_types specified in MDMC the integers used should be monotonically increasing.
[ ]:
# from MDMC.readers.configurations import read
atoms = configurations.read('data/Paracetamol.cif', atom_type=[1, 2, # Oxygens
3, # Nitrogen
4, 5, 6, 7, 6, 6, 6, 8, # Carbons
9, 9, 9, 10, 10, 10, 10, 11, 12]) # Hydrogens
paracetamol = Molecule(atoms=atoms)
# Print the total number of bonds and the number of unique bonds
bonds = list(filter(lambda x: x.name == 'Bond', paracetamol.interactions))
print('Number of bonds: {}'.format(len(bonds)))
print('Number of unique bonds: {}'.format(len(set(bonds))))
Equivalence specified by names
The names must be strings which are only shared by equivalent atoms. These set the name attribute for each Atom. As covered in the guide on applying a force field, if each Atom has an Atom.name which is defined in a ForceField, the ForceField can be applied to set the Parameter values for all interactions. An example of this is shown below, where the names provided are the OPLSAA atom types:
[ ]:
# from MDMC.readers.configurations import read
atoms = configurations.read('data/Paracetamol.cif', name=['109', '177', # Oxygens
'207', # Nitrogen
'208', '108', '90', '178', '90', '90', '90', '185', # Carbons
'85', '85', '85', '91', '91', '91', '91', '183', '110']) # Hydrogens
paracetamol = Molecule(atoms=atoms)
# Print the total number of bonds and the number of unique bonds
bonds = list(filter(lambda x: x.name == 'Bond', paracetamol.interactions))
print('Number of bonds: {}'.format(len(bonds)))
print('Number of unique bonds: {}'.format(len(set(bonds))))
The paracetamol can now be added to a Universe and the OPLSAA ForceField can be applied to set the Parameter values:
[ ]:
from MDMC.MD import Universe
universe = Universe(10.)
universe.add_structure(paracetamol)
universe.add_force_field('OPLSAA')
[ ]:
print(universe)
[ ]:
universe.nonbonded_interactions
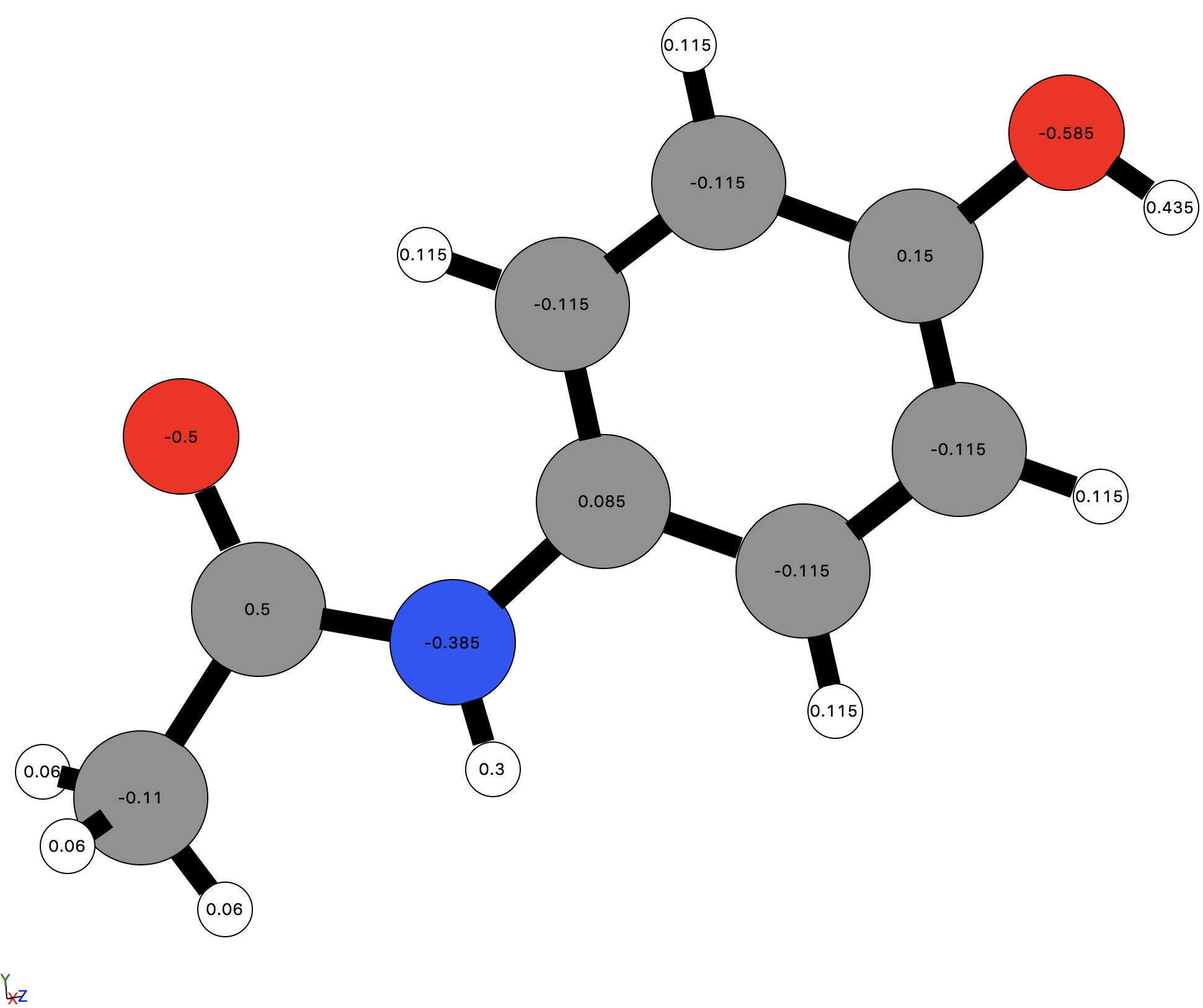
The effect of this can be seen if we visualize the paracetamol molecule using the 'ASE' viewer and select View -> Charges from the GUI menu bar.
[ ]:
view(paracetamol, viewer='ASE')
Getting configurations directly from databases or PubChem
As configurations are read via ASE, MDMC supports converting any ASE Atoms object into a list of MDMC Atoms. Indeed, all our file reader is really doing is this:
ase_atoms = ase.io.read(file)
atoms = MDMC.MD.ase.convert.ASE_to_MDMC(atoms)
and much in the same way, anything that can be read as ASE atoms can be converted into an MDMC Molecule. In particular, we will see how to read in molecules from ASE’s built-in databases and from PubChem.
A warning on using databases and the Internet: For quality of research, it is recommended that a file is used (and made available) for any read-in configurations used in research. This allows both the creator of the configuration to be credited, and also ensures your work is reproducible (as databases or websites can change!)
Building from an ASE built-in database
ASE has several built in databases containing many popular molecules. The ase.build.molecule function allows users to read in ASE molecules from these databases.
[ ]:
from ase.build import molecule
from MDMC.MD.ase.convert import ASE_to_MDMC
# we read in a benzene molecule
ase_atoms = molecule("C6H6")
MDMC_atoms = ASE_to_MDMC(ase_atoms)
benzene = Molecule(atoms=MDMC_atoms)
view(benzene)
Please see the relevant documentation for more information on ASE’s built-in databases.
Building from the Internet
ASE also has an integration with PubChem, which allows you to search the PubChem website directly and read in the result as an ASE Atoms object. This can again be easily converted to an MDMC molecule.
[ ]:
from ase.data.pubchem import pubchem_atoms_search
# search by name, CID or SMILES string
ase_aspirin = pubchem_atoms_search(name='aspirin')
aspirin = Molecule(atoms=ASE_to_MDMC(ase_aspirin))
ase_glucose = pubchem_atoms_search(cid='5793')
glucose = Molecule(atoms=ASE_to_MDMC(ase_glucose))
ase_benzaldehyde = pubchem_atoms_search(smiles='C1=CC=C(C=C1)C=O')
benzaldehyde = Molecule(atoms=ASE_to_MDMC(ase_benzaldehyde))
[ ]:
view(aspirin)
[ ]:
view(glucose)
[ ]:
view(benzaldehyde)